Research Program - Epigenetic mechanisms and genome editing tool development
Cytosine DNA methylation is an epigenetic modification of DNA that is usually associated with the stable and heritable repression of gene transcription. It is particularly important in the silencing of transposable elements, which can otherwise transpose and cause deleterious mutations and DNA damage. DNA methylation is also involved in the regulation of protein coding gene expression, which contributes to development, imprinting, and X-chromosome inactivation. DNA methylation can in some cases also cause gene activation.
We study DNA methylation in the model plant Arabidopsis thaliana because of its facile genetics, small size, and trim genome. Furthermore, unlike other organisms like mouse, where DNA methylation mutants are inviable, Arabidopsis can tolerate mutations that virtually eliminate methylation, allowing for further study. Arabidopsis methylation mutants display developmental abnormalities because of defects in the methylation of key genes that regulate development. Many of these methylation differences can be inherited in sexual progeny, giving rise to epigenetic alleles of genes. For example, the FWA gene can adopt two stable epigenetic states, either methylated and silent, or unmethylated and active causing a later flowering phenotype (figure below). The underlying DNA sequences of these epialleles are identical, so the only difference is the methylation state and expression state. Remarkably, both the methylated/silent state and the unmethylated/active state are very stably inherited in progeny plants for many generations.
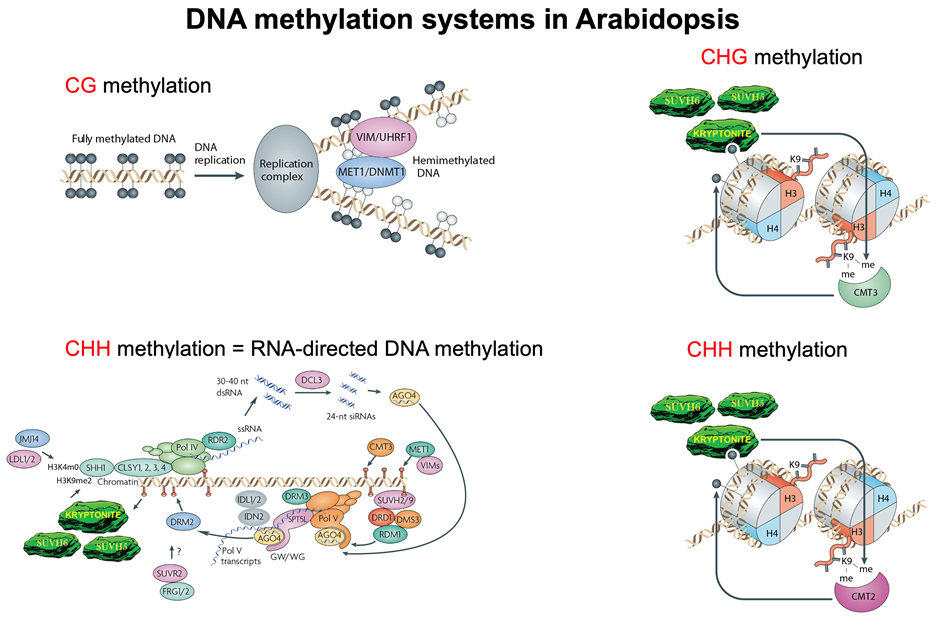
We have taken advantage of these stable DNA-methylation based epialleles to perform genetic screens for mutants affecting DNA methylation. These studies have revealed that DNA methylation is controlled by 1) the specificity of several different DNA methyltransferases, 2) targeting by other chromatin modifications such as histone methylation, and 3) targeting of specific DNA sequences by small interfering RNAs (siRNAs) and longer non-coding RNAs. In addition to genetics, we rely heavily on computational biology and biochemistry in our work. We also have a smaller mammalian program in the lab, focused on studies of conserved regulators of DNA methylation. The figure below represents our current view of the four different pathways that are involved in the establishment and maintenance of DNA methylation in three different cytosine sequence contexts, CG, CHG, and asymmetric CHH sites (H = A, T, or C).
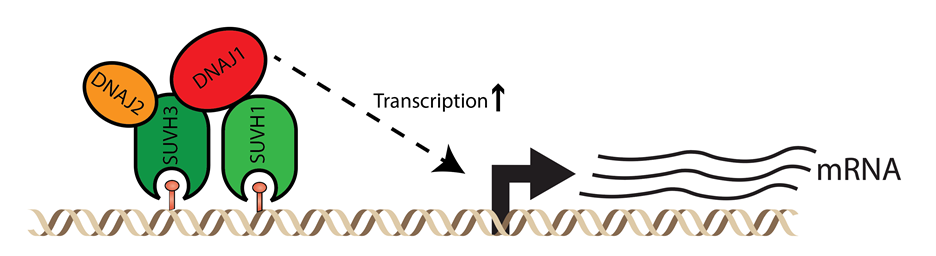
Although we now understand the mechanisms of DNA methylation initiation and maintenance in some detail, we still have a very poor understanding of how DNA methylation is read and translated into a signal for regulating gene expression. One current focus of the lab is to study these processes that act downstream of DNA methylation. For example, we have recently discovered a protein complex that binds to sites of RNA-directed DNA methylation and causes upregulation of protein coding genes that are in close proximity to the DNA methylation (figure below). This complex therefore counteracts the normally repressive effect of transposon insertion near genes by boosting expression of these genes. The mechanism of stimulation of gene expression by this complex is as yet unknown.
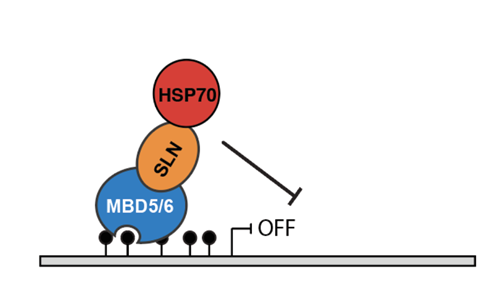
We also discovered a complex of proteins that is involved in repression of gene expression by DNA methylation. The complex consists of two methyl binding domain proteins called MBD5 and MBD6 that bind to genomic sites with dense CG DNA methylation and form a complex with a J-domain protein we named SILENZIO. Both the mbd5 mbd6 double mutants and silenzio mutants show derepression of methylated transposons and genes. SILENZIO acts in part via its interaction with Hsp70 proteins, suggesting that molecular chaperone activity regulates gene silencing (figure below). We are currently trying to understand the mechanism of repression by this complex.
We are also studying a number of additional new complexes that act downstream of DNA methylation to control gene expression, as well as studying the MORC proteins that we previously found to act downstream of DNA methylation.
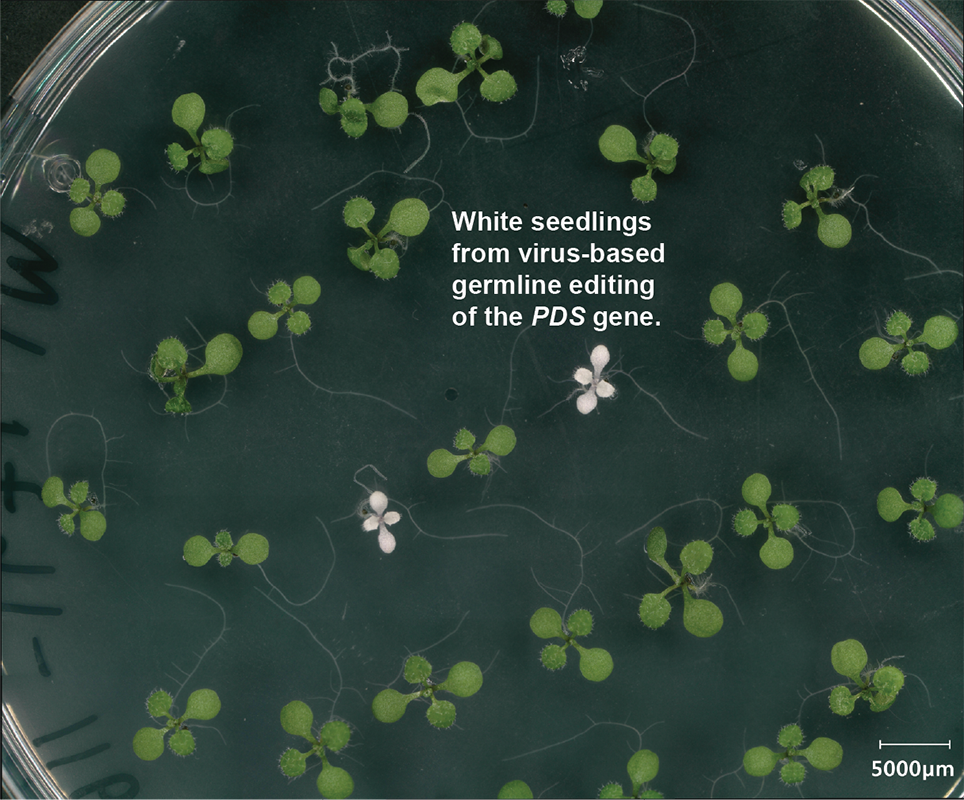
We also have a newer project in the lab aimed at developing tools for genome editing of plant genomes using plant RNA viruses. RNA viruses are interesting vehicles for delivering proteins and RNAs to plants given their natural ability to replicate and spread throughout plants. They replicate purely by RNA and therefore do not leave traces of themselves in plant genomes. They also can gain access to the germline of plants and yet are themselves restricted from passing to future generations. Because of the limited cargo capacity of these viruses, it has not been possible to encode common CRISPR systems like Cas9 inside of them. To solve this problem, we have been working with Jennifer Doudna’s lab at UC Berkeley to screen for and engineer tiny CRISPR systems that are small enough to be encoded in plant viruses. Our first breakthrough in this work was recently published (Nature Plants 2025), in which we showed that a modified tobacco rattle virus could cause germline editing in Arabidopsis. Current projects are aimed at improving and augmenting these systems, so they may be someday used for plant CRISPR screens as well as engineering of crop plants.